Clinical features
- It accounts for around 50% of all childhood soft-tissue sarcomas
- 1/3 of the cases are diagnosed in the first 3 years of life
- Can be congenital
- Most arise from skeletal muscle, although they may also arise in viscera
- Those arising from skeletal muscle are particularly associated with genetic fusions
- About 65% are diagnosed in children (50% occur in the first decade)
- Slight male predominance
- Rare familial forms are reported in association with Li-Fraumeni, basal cell nevus syndrome, pleuropulmonary blastoma, Beckwith-Wiedemann syndrome and neurofibromatosis
- Association with congenital anomalies of the central nervous system, genitourinary tract, gastrointestinal tract and cardiovascular system
- Association with low economic background
- Bimodal age distribution: peak at 3 – 5 years; and at 16 -17 years
- 95% of the cases in children belong to the alveolar or embryonal subtype
- Clustering of the primary tumour site, age and morphology (embryonal/alveolar) is a distinctive feature:
- Embryonal
- In infants (may be congenital)
- Mainly located in the orbit or perineum
- Head and neck, nasopharynx and genitourinary tract
- Rarely spreads to regional lymph nodes
- Alveolar
- In adolescence
- The most aggressive subtype
- Extremities, perineal and periorbital regions
- Uncommon presentation: leukaemia-like
- Metastasis to regional lymph nodes and along fascial planes
- Embryonal
- Symptoms depend on the localization
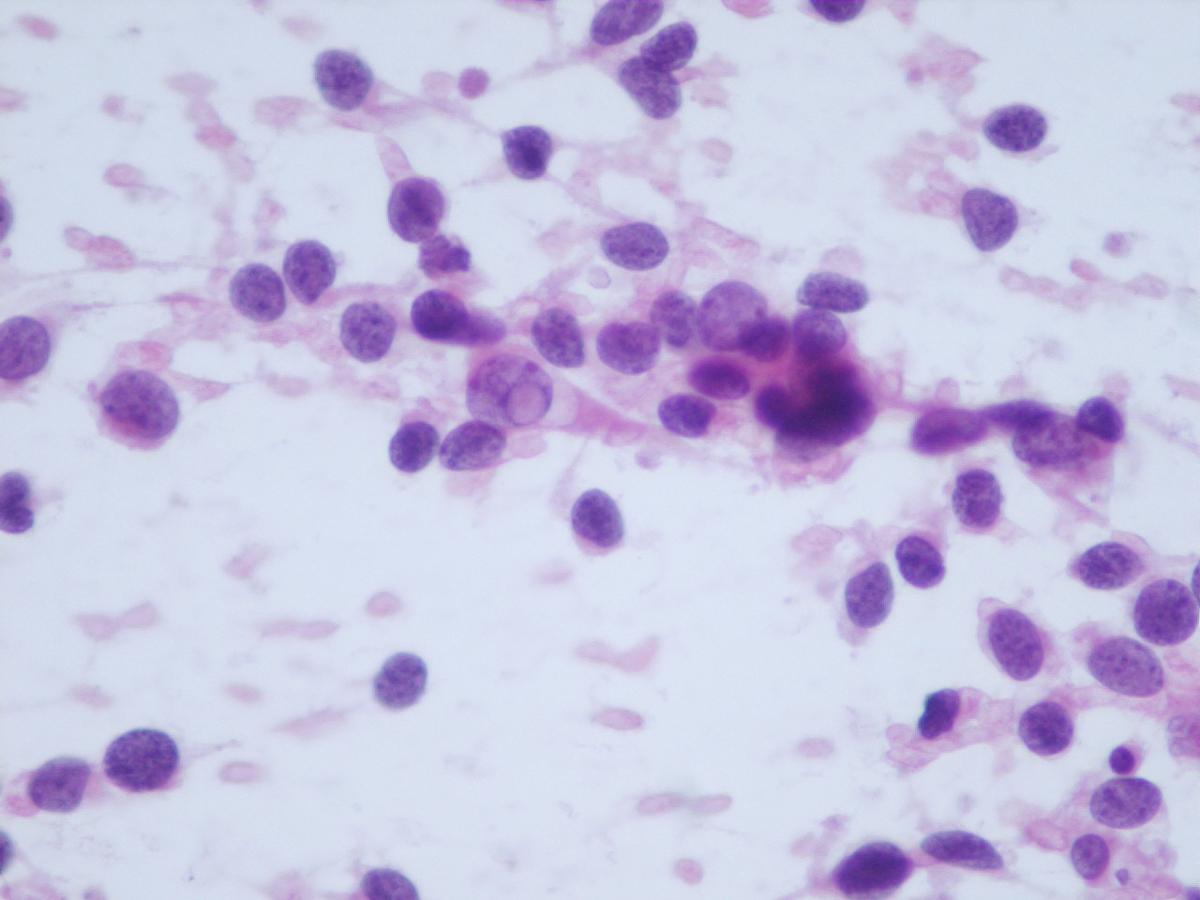
Fig 13a – Alveolar rhabdomyosarcoma – Cellular smears with a background with apoptotic bodies and loosely cohesive aggregates of uniform, small round blue cells (H&E).
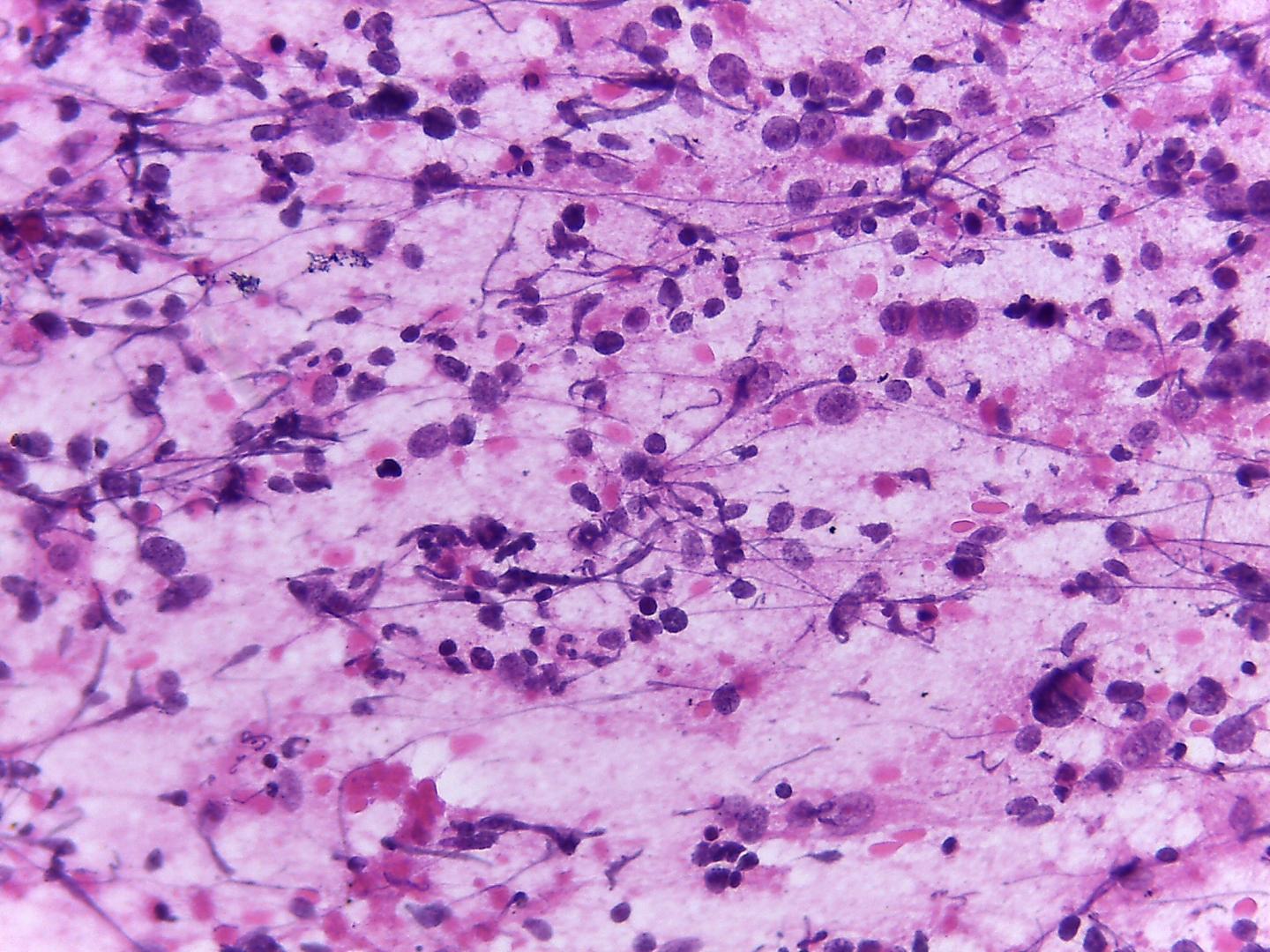
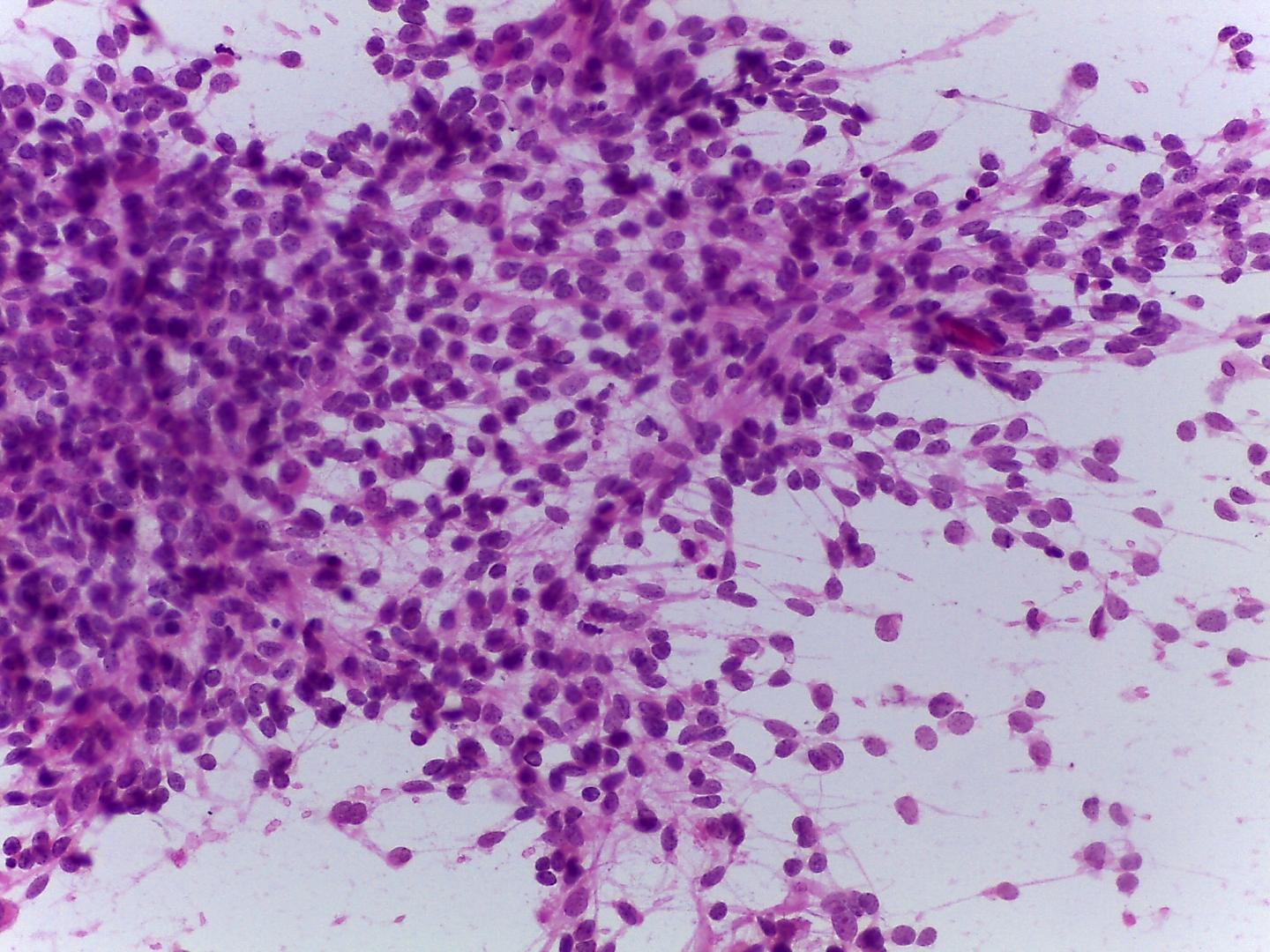
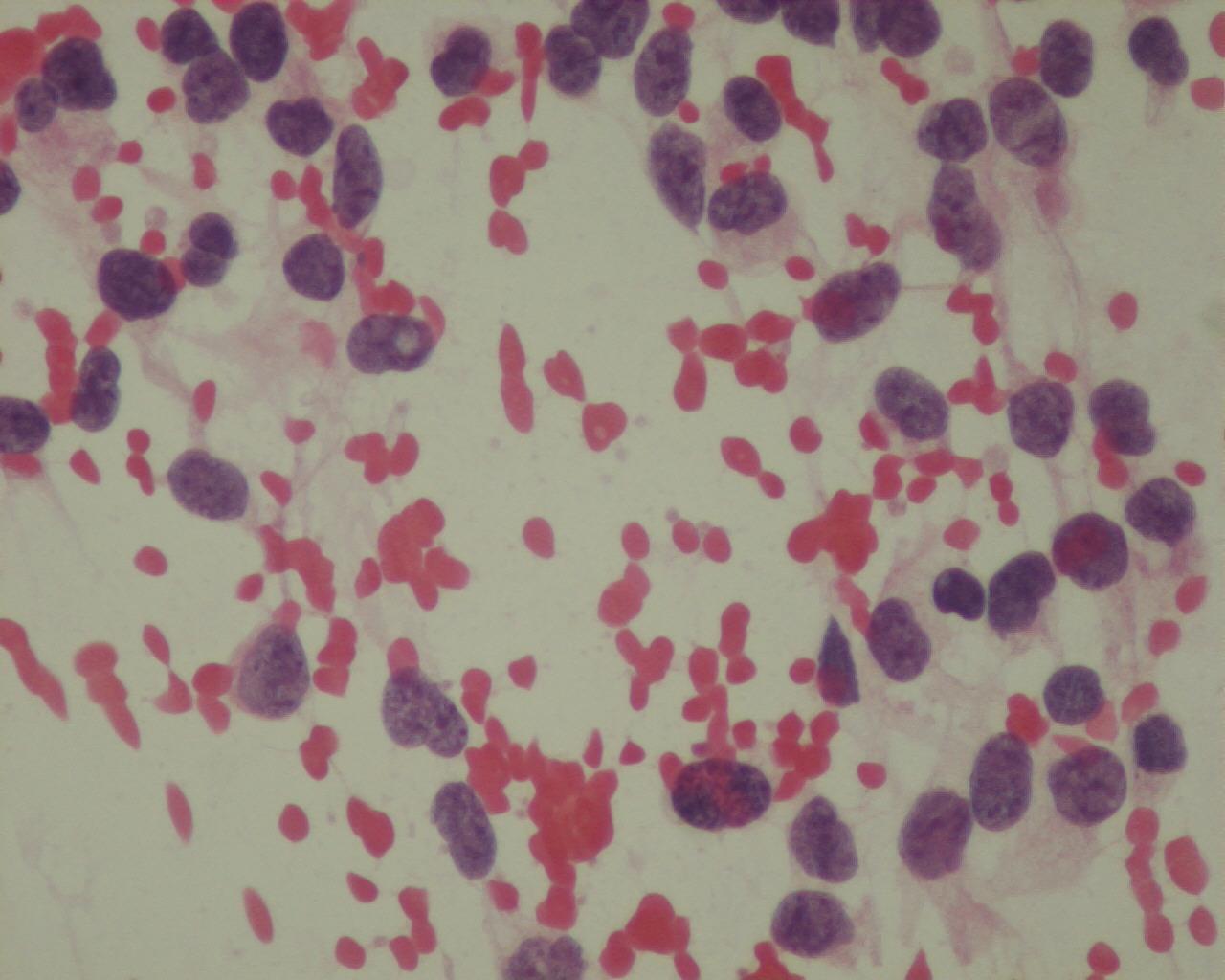
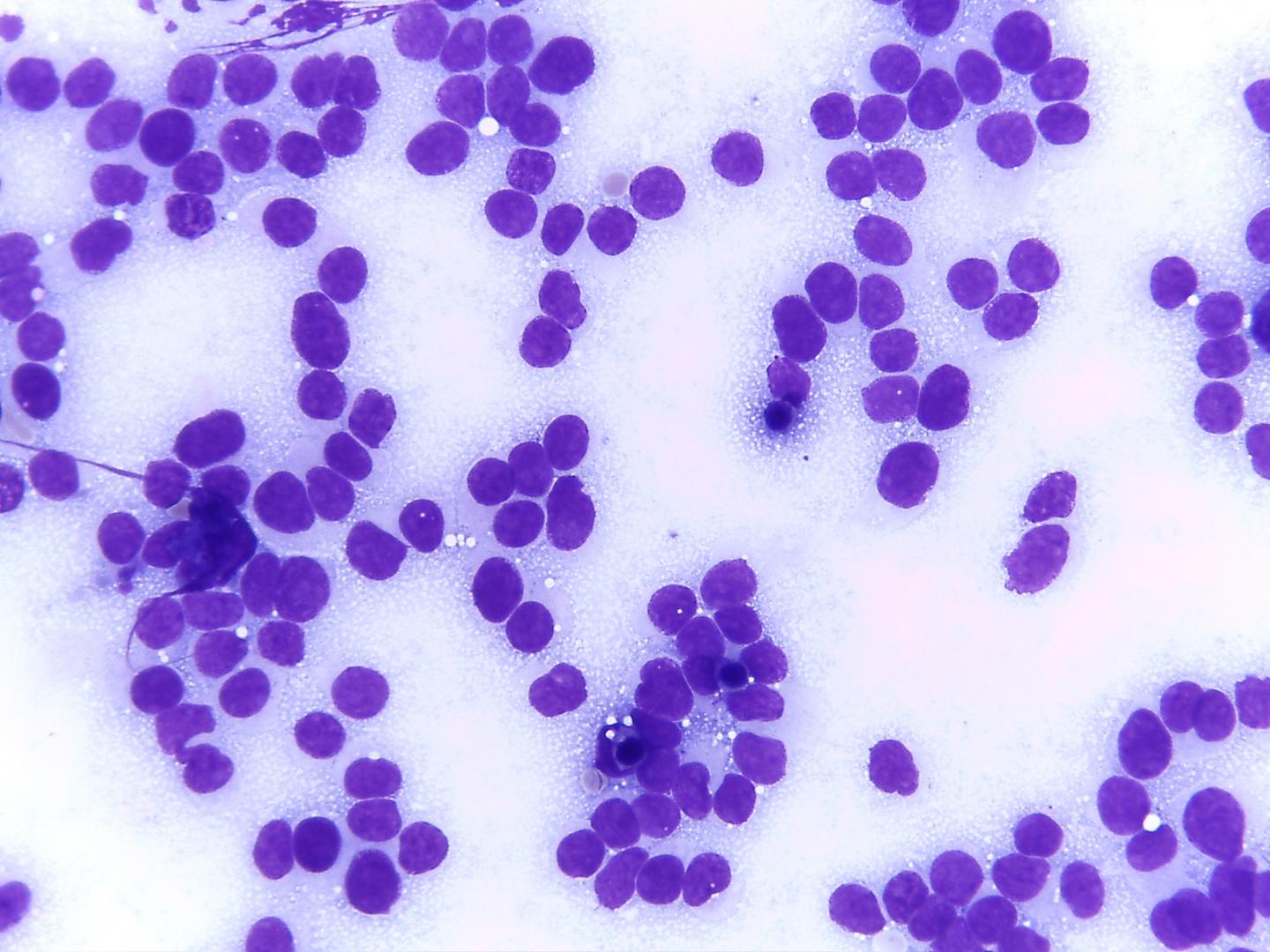
Fig 14a– Alveolar rhabdomyosarcoma – Giemsa- remark the typical tigroid background
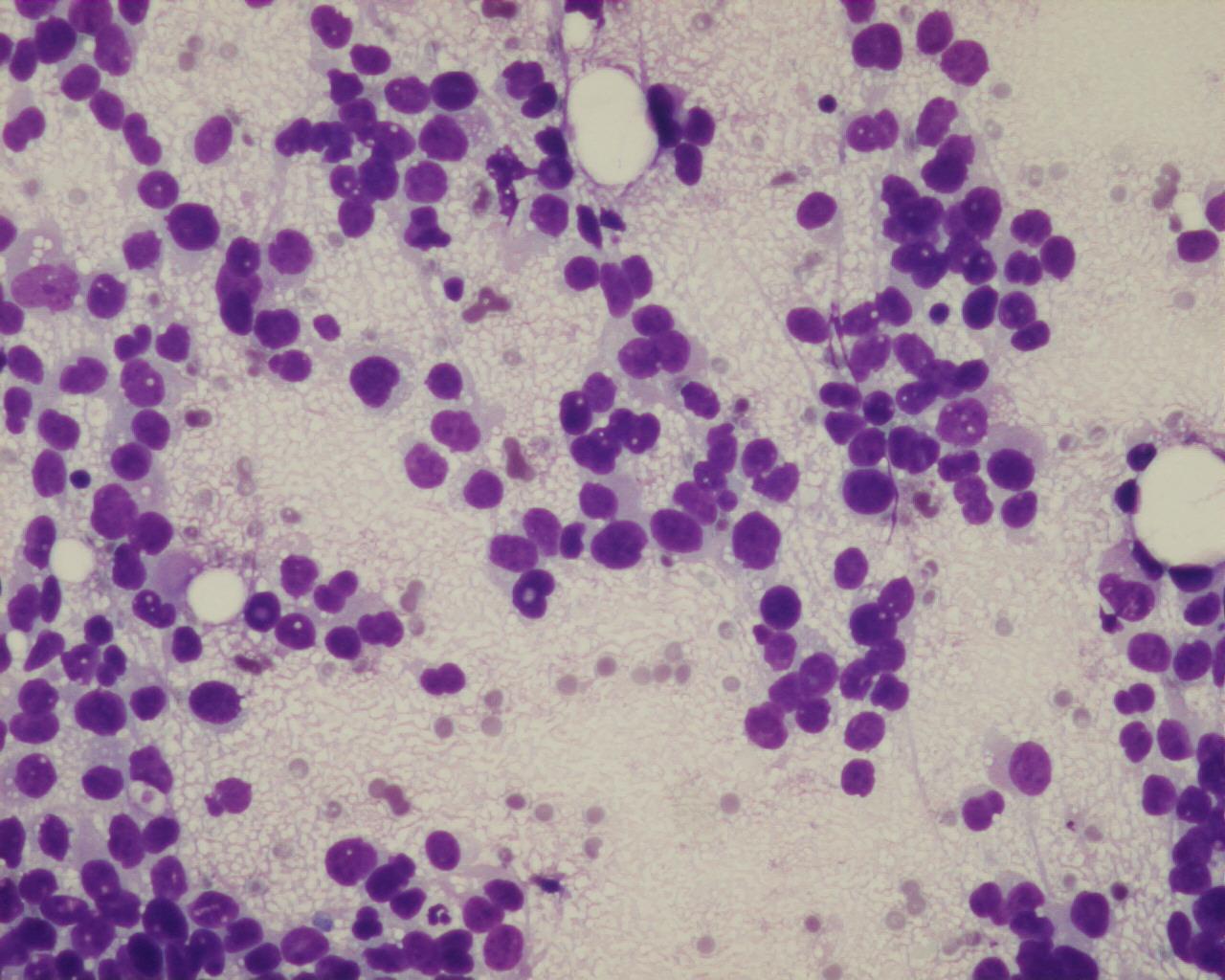
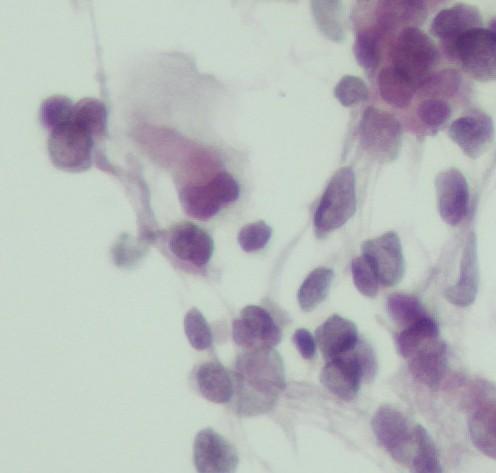
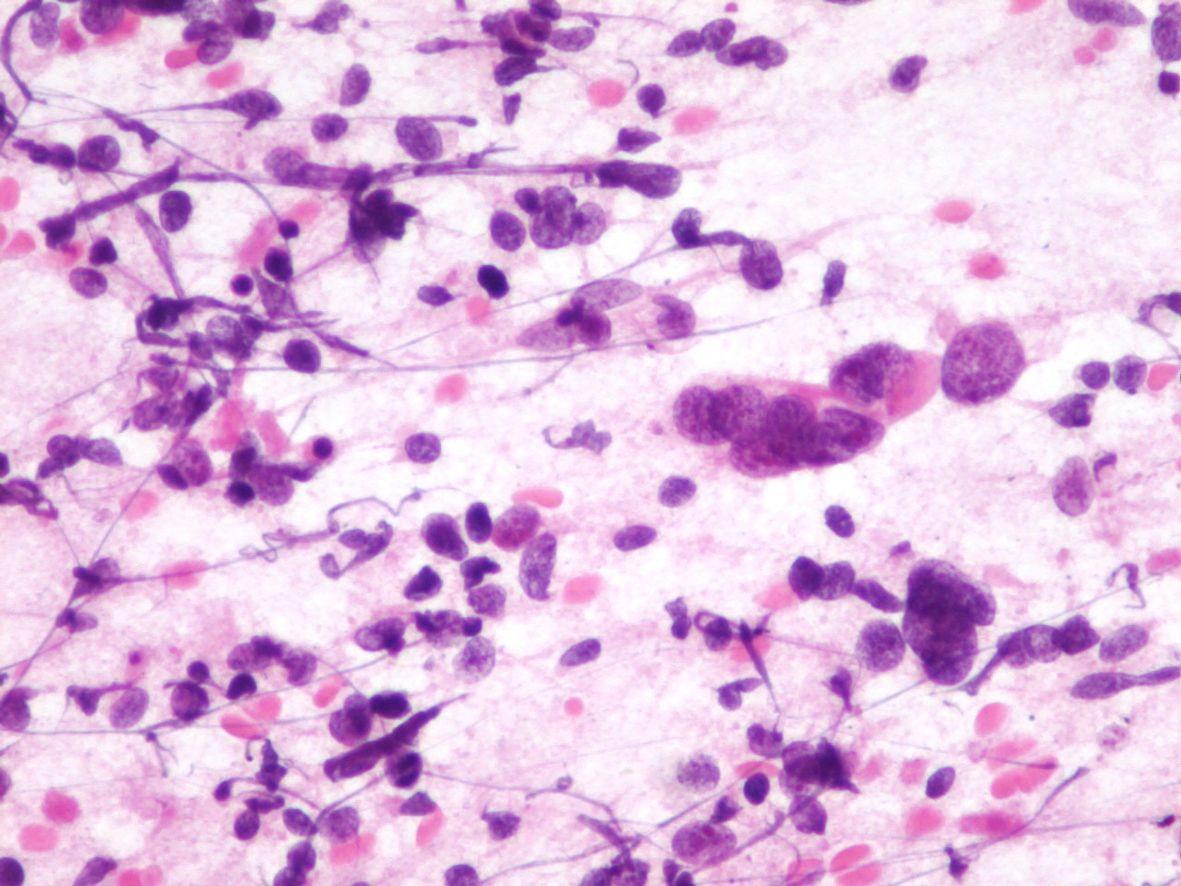
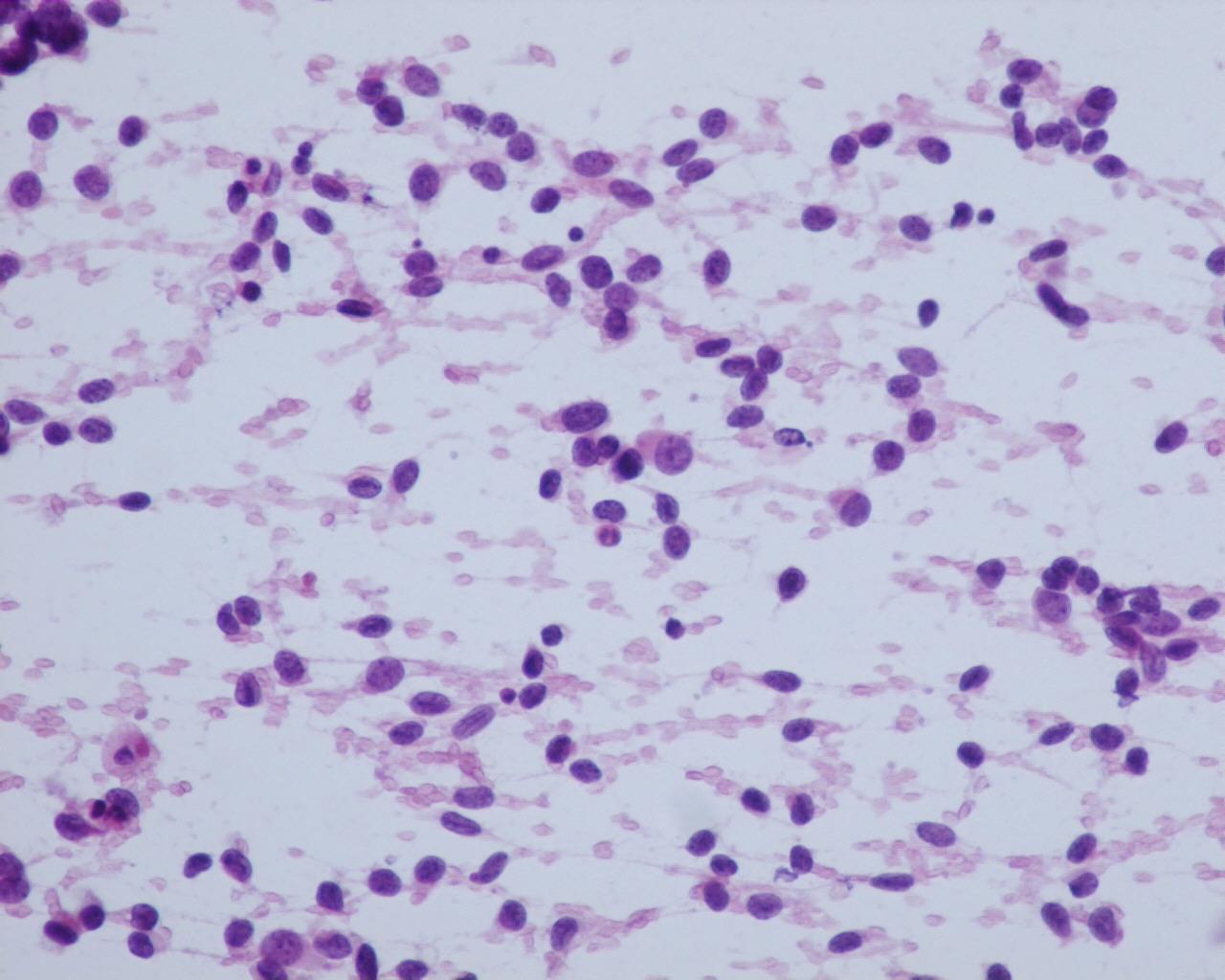
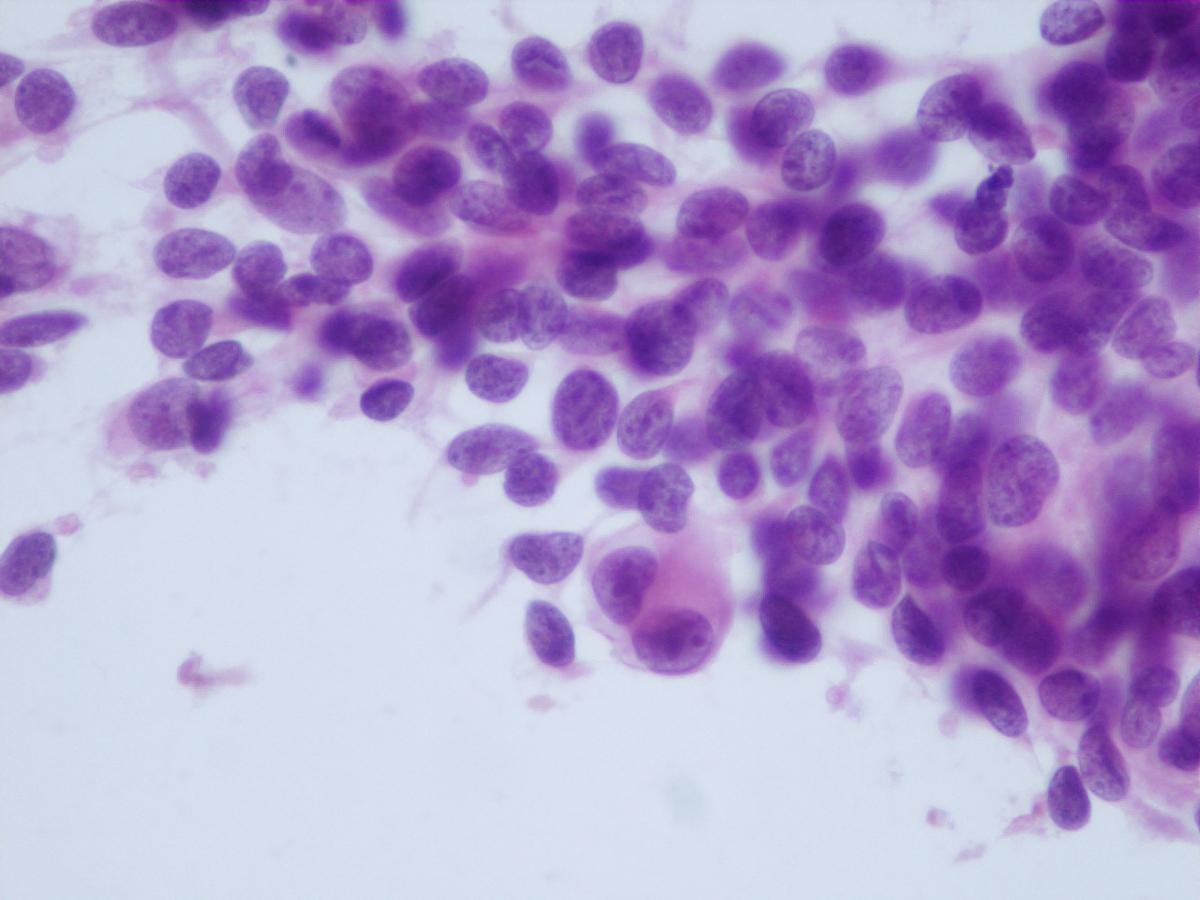
Fig 16 a- Embryonal rhabdomyosarcoma- Nuclei can have pseudo inclusions (H&E)
- Embryonal (ERMS)
- Clusters or isolated cells
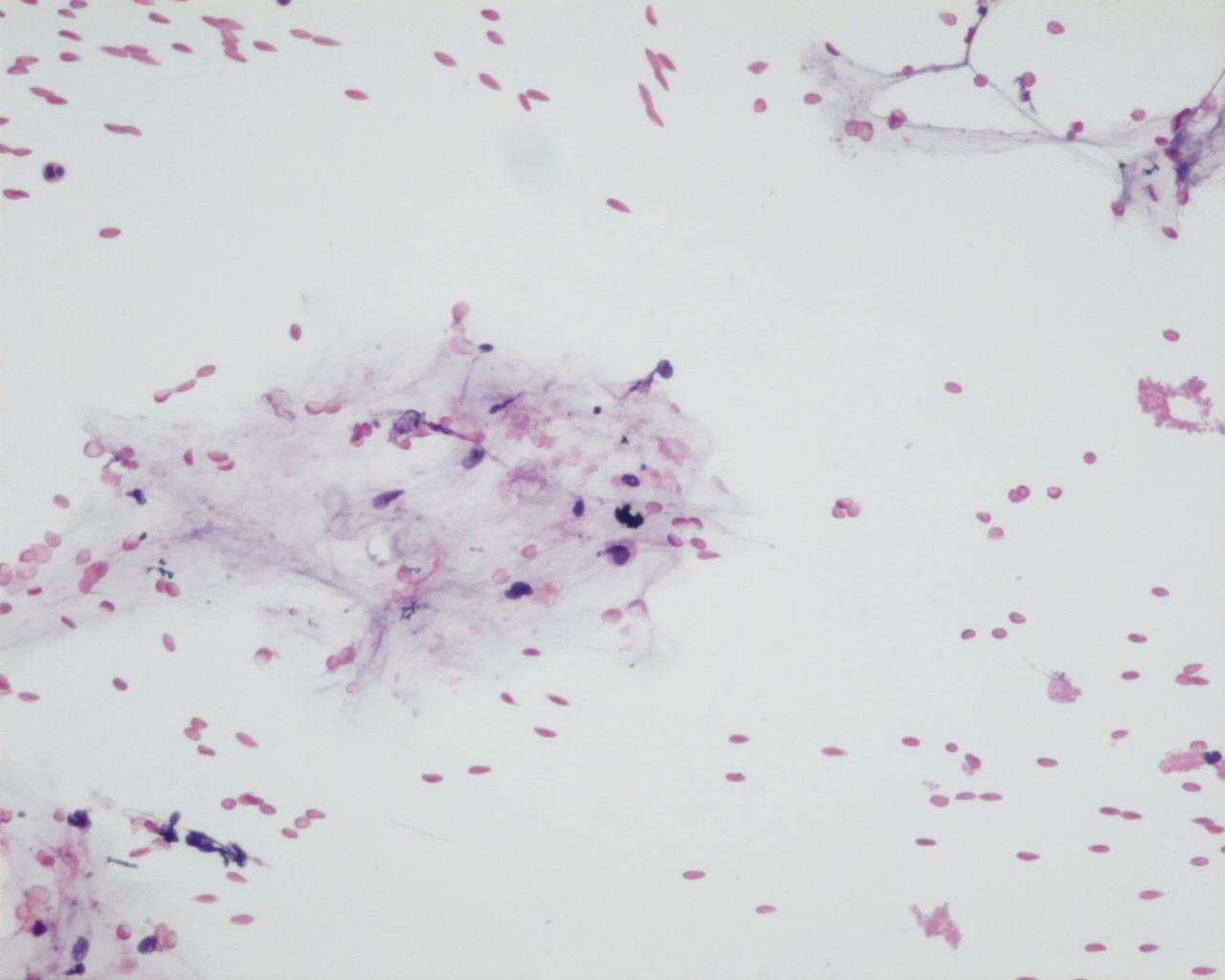
- Myxoid stroma and “tigroid” background in 20% of the cases (Giemsa staining)
- Lymphoglandular-like bodies present in 20%
- More cellular variation: whole range from primitive mesenchymal cells (fusiform or stellate cells) to highly differentiated rhabdomyoblasts
- Binucleated or multinucleated tumour cells (strap cells) provide an important clue for differential diagnosis with other entities
- Presence of “tadpole” or “racket cells” and ribbon-like cells
- Nuclei with finely granular chromatin
- Cytoplasmic glycogen vacuoles
- Alveolar (ARMS)
- Cellular smears
- Background with apoptotic bodies can simulate a Burkitt cell lymphoma
- Tigroid background
- Loosely cohesive aggregates of uniform, small round blue cells
- Fine chromatin
- Multinucleated neoplastic giant cells with eosinophilic cytoplasm
- Inconspicuous nucleoli (occasionally can be prominent)
- Vacuoles of glycogen in the cytoplasm
- In some cells, a rhabdoid phenotype can be seen
- Although these two main subtypes of rhabdomyosarcoma show some differences in cytomorphology, most authors do not rely on cytology alone, to make the differential diagnosis
- Cytogenetic and molecular techniques together may be of great help; confirmation by means of histology is sometimes required
Immunocytochemistry
- Alpha-actin: positive
- Desmin: positive
- Myosin: positive
- Myoglobin: positive
- Myogenin: positive (more sensitive than Myo-D1 on formalin-fixed material); strong and diffuse nuclear expression mainly in alveolar type and correlated with poor prognosis
- MyoD1: positive – (more sensitive than myogenin in frozen material). Together with myogenin these are the more sensitive and specific markers of skeletal muscle
- HHF35: positive
- CD56 (N-CAM): positive , sometimes strongly positive (11,12)
- “Aberrant” staining: NSE, synaptophysin, Leu7, cytokeratin, neurofilaments, S-100 protein and CD99
- WT1: Positive (only cytoplasm)
Genetic studies
- Alveolar subtype has two main characteristic translocations:
- t (2; 13) (q35; q14) in 70% of the cases
- t(1;13)(p36;q14) in 10-20% of the cases
- Associated with:
- younger patients
- better prognosis
- involvement of extremities
- Associated with:
- In 30% of the cases no translocation is found by RT_PCR
- Solid variants are more likely to be PAX/FKHR negative
- Fusion negative RMS behave similarly to ERMS
- Embryonal subtype :
- Loss of heterozygosity (LOH) at the 11p15 locus of the IGF II gene
- 1p deletion
- Both subtypes of rhabdomyosarcoma have over expression of the IGF II gene
Differential diagnosis
- Peripheral neuroectodermal tumours (PNET)
- Tigroid background
- Synaptophysin: positive
- Chromogranin: positive
- NSE: positive
- Desmin: positive (occasional)
- Myogenin or actin: negative
- t(11;22)(q24;q12)
- Desmoplastic small cell tumour
- Usually intra-abdominal
- Nuclei with granular chromatin
- Higher C/N ratio
- Prominent desmoplastic stroma
- Divergent differentiation
- Myo-D1-negative
- Myogenin: negative
- WT1- nuclear positivity
- Neuroblastoma
- Usually secrete catecholamine’s
- Neuroblasts generally at different stages of differentiation
- Typical neuroendocrine chromatin (“salt-and-pepper”)
- Neuropil frequently present
- Rosettes frequently present
- CD56 N-CAM: positive
- Vimentin: negative
- Synaptophysin: positive
- Muscle markers: negative
- Lymphoma
- Lymphoglandular bodies (Giemsa stain)
- Single cells
- CD45: positive
- Muscle markers: negative
- Rhabdoid tumour
- Lack of strap-shaped and ribbon-like cells
- More monotonous population with rhabdoid features
- Pale nuclei with vesicular chromatin
- INI1 : negative
Main points
- Overall five-year survival: 50%-75%
- Prognosis depends on:
- Age at diagnosis; best survival is found when diagnosed between one and eight years of age
- Tumour site: two-year disease-free survival in orbital region is 77%, versus 24% for intrathoracic tumours
- Stage
- International prognostic classification of paediatric rhabdomyosarcoma
- Superior prognosis
- Botryoid
- Spindle
- Intermediate prognosis
- Embryonal
- Poor prognosis
- Alveolar
- RMS with diffuse anaplasia
- Undifferentiated sarcoma
- Uncertain prognosis
- Rhabdoid features
- Superior prognosis
- t (1; 13) (q36; q14)
- associated with younger patients
- and tumours in the extremities