Clinical features
- Rare tumour that accounts for less than 2% of paediatric renal tumours
- 60% are diagnosed in the first year of age
- Striking clinical associations:
- Hypercalcemia in 15%,- associated with tumour production of parathormone and parathormone-like substances
- 15% of synchronous or metachronous genetically independent primary embryonal brain tumour (PNET–like neoplasm)
- Often compromises the renal hilum at presentation
- No relation with Wilms’ tumour or any syndrome
- First description in the kidney; it is now well-documented in other locations such as liver, soft tissues, brain, genitourinary tract and skin
- Often metastasized at presentation (bone, brain and regional lymph nodes)
- High-risk tumours (SIOP-2001 revised working classification of renal tumours of childhood)
Fig 7a (H&E) – Rhabdoid tumor- Round to polygonal rhabdoid dispersed cells in a background of necrosis. Eccentric nuclei with an intracytoplasmic eosinophilic inclusion can be seen
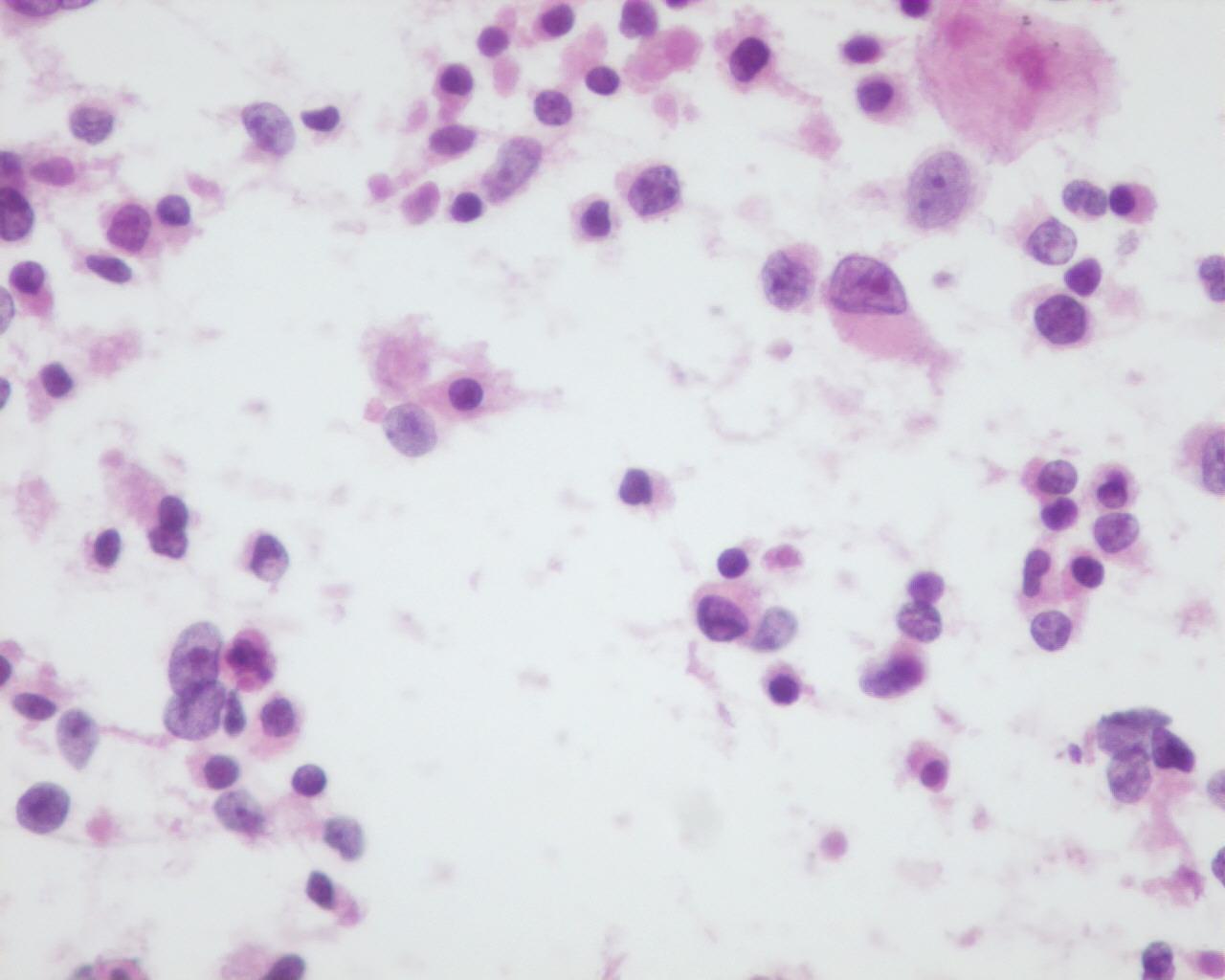
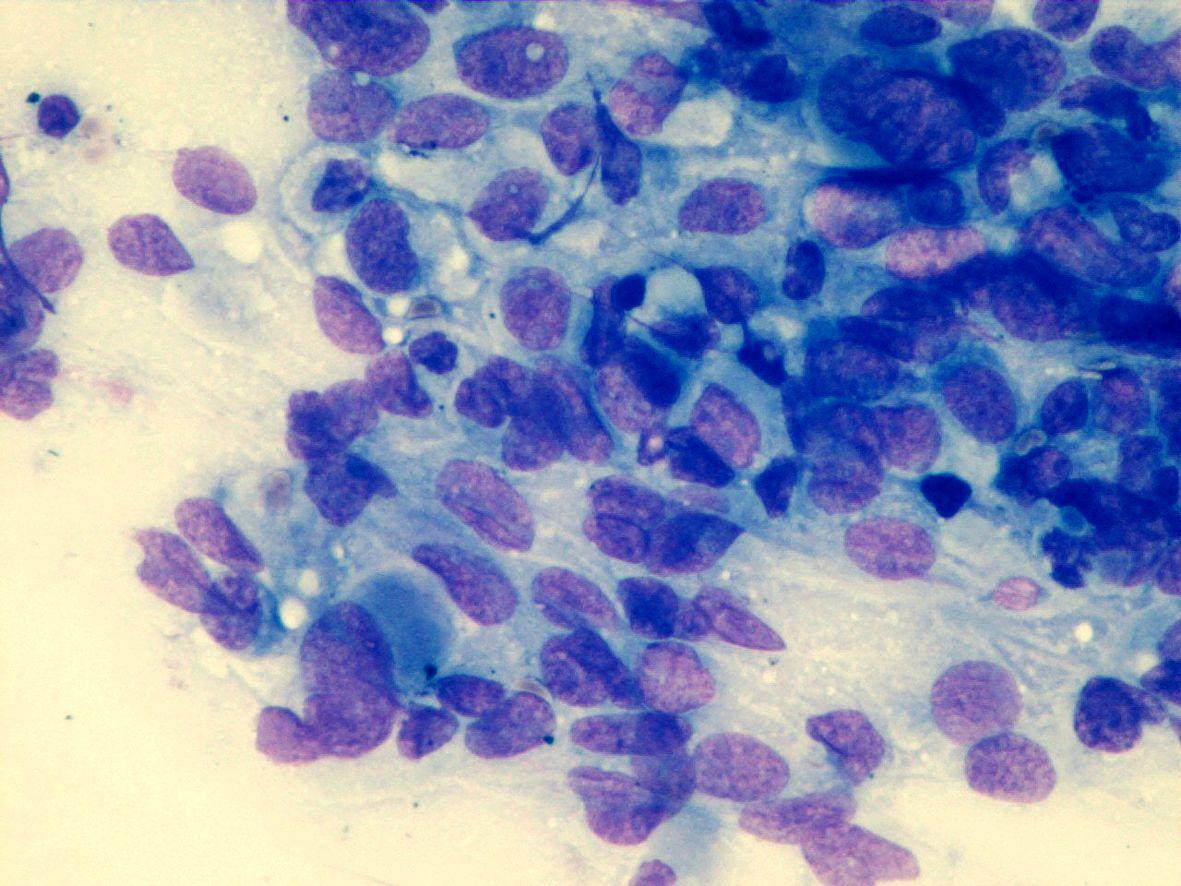
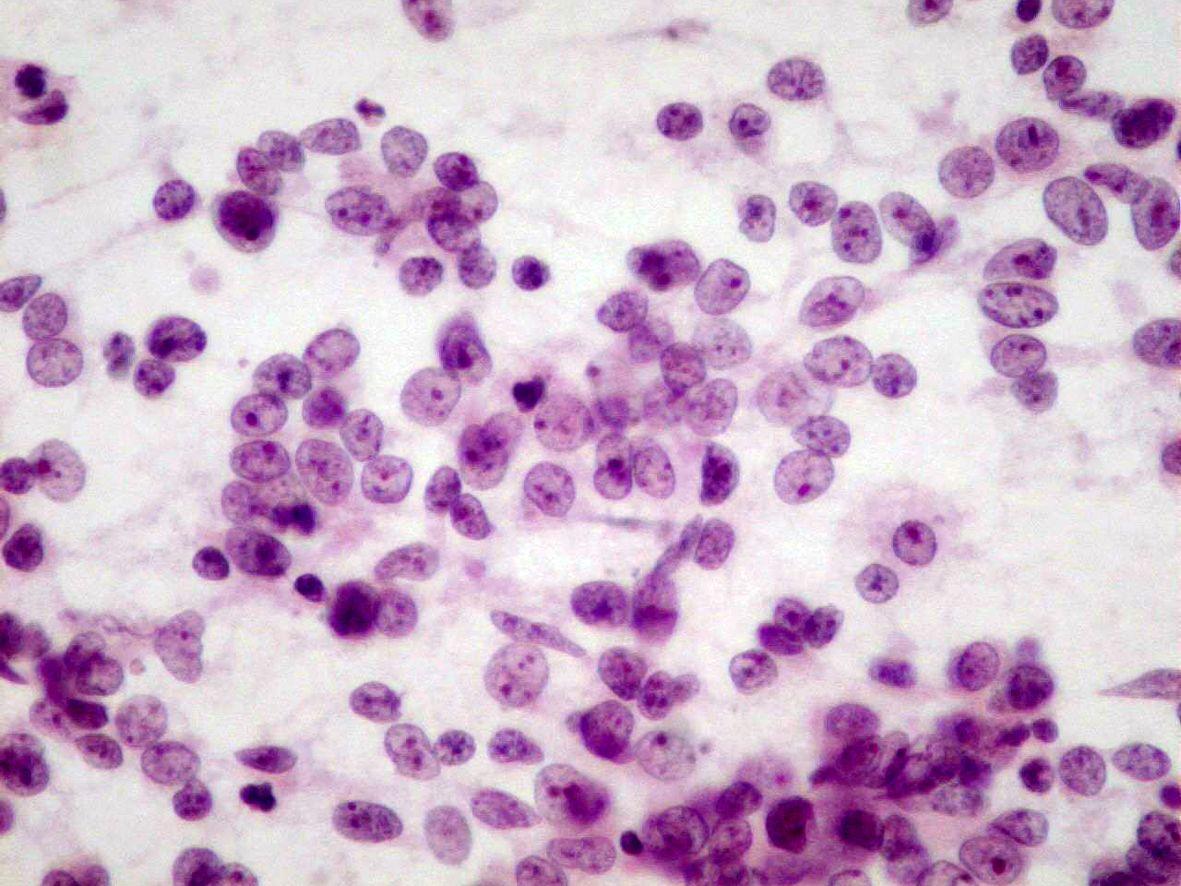
- Round to polygonal rhabdoid-like dispersed cells or in clusters:
- Bare nuclei
- Cell with irregular nuclei
- Reticular chromatin
- Single eosinophilic macronucleoli
- Clear area around the macronucleoli
- Fragile, pale, and eccentric cytoplasm
- Intracytoplasmic eosinophilic inclusions
Immunocytochemistry
| Vimentin: positive (dot)Cytokeratin: positive (dot)EMA: positive | Coexpression |
| Desmin: rarely positiveCD99: positiveSynaptophysin: positive (variable)CD10:positiveMyogenin: negativeSmooth muscle actin: negativeCD56(NCAM): negativeCD57: negativeINI1: negative-loss of nuclear positivity (most other tumours with rhabdoid features have detectable INI1 protein). |
Genetic studies:
Variable findings:
- Frequent monosomy 22
- Deletion at 22q11 in chromosome 22
- Biallelic inactivation of the tumour suppressor gene locus of hSNF5/INI1 gene
- Association with alterations on the short arm of chromosome 11
- Patients with germline mutation of HSNF5/INI1 are more prone to have malignant embryonal tumours of posterior fossa
Differential Diagnosis
- Nephroblastoma, predominantly blastemal
- Regular nuclei with fine chromatin
- Inconspicuous nucleoli
- No cytoplasmic inclusions
- Absence of coexpression of vimentin and cytokeratin’s
- INI 1 : positive
- Rhabdomyosarcoma
- Anisokaryosis
- Trap cells
- Tigroid background
- Desmin: strongly positive
- Myogenin: positive
- INI 1 : positive
- Rhabdomyoblastic differentiation on electron microscopy
- Neuroblastoma
- Nuclei with salt-and-pepper chromatin
- Neuroblasts in different stages of maturation
- Fibrillary matrix
- Cytokeratin: negative
- Vimentin: usually negative
- Renal PNET
- Most common in young adults
- Nuclei with coarse chromatin
- CD99: positive
- INI1: positive
- t (11:22) (q24; q12)
- Renal cell carcinoma
- Rarely affects children under the age of two years
- High degree of cellular cohesion
- Absence of intermediate filaments (electron microscopy)
- Lymphoma
- Lymphoglandular bodies
- CD45: positive
- Cytokeratin: negative
Main points
- Initially described in the kidneys by Beckwith and Palmer as a possibly sarcomatous variant of Wilms’ tumour
- Later described in soft tissues and other organs
- Most likely of primitive epithelial derivation
- Highly aggressive tumour:
- 82% with metastases at presentation
- 90% of patients die within two years
- Age affects prognosis: age under six months of age –poor prognosis; age older two years of age have better prognosis
- Occur in patients with germline mutations in hSNF5/INI1
- A familial “rhabdoid predisposition syndrome” has been described in families with constitutional inactivating mutations of hSNF5/INI1.