linical features
- Malignant embryonal tumour
- Common type of paediatric cancer (63% of paediatric cancers)
- 95% of all paediatric renal tumours
- 5-10% are bilateral
- Rarely as an extrarenal tumour
- 1-2% have familial origin
- Uncommon in neonates; peak of incidence between three and four years; usually diagnosed before 6 years of age.
- Female predominance (unlike the majority of childhood cancers)
- Palpable abdominal mass with pain (generally due to necrosis) is the most common presentation
- Hypertension and haematuria
- Associated with hereditary conditions such as WAGR syndrome (Complete deletion of WT1 gene), Beckwith-Wiedemann syndrome (loss heterozygoty of chromosome 11p), hemi hypertrophy, Denys-Drash syndrome (point mutation of WT1 gene).
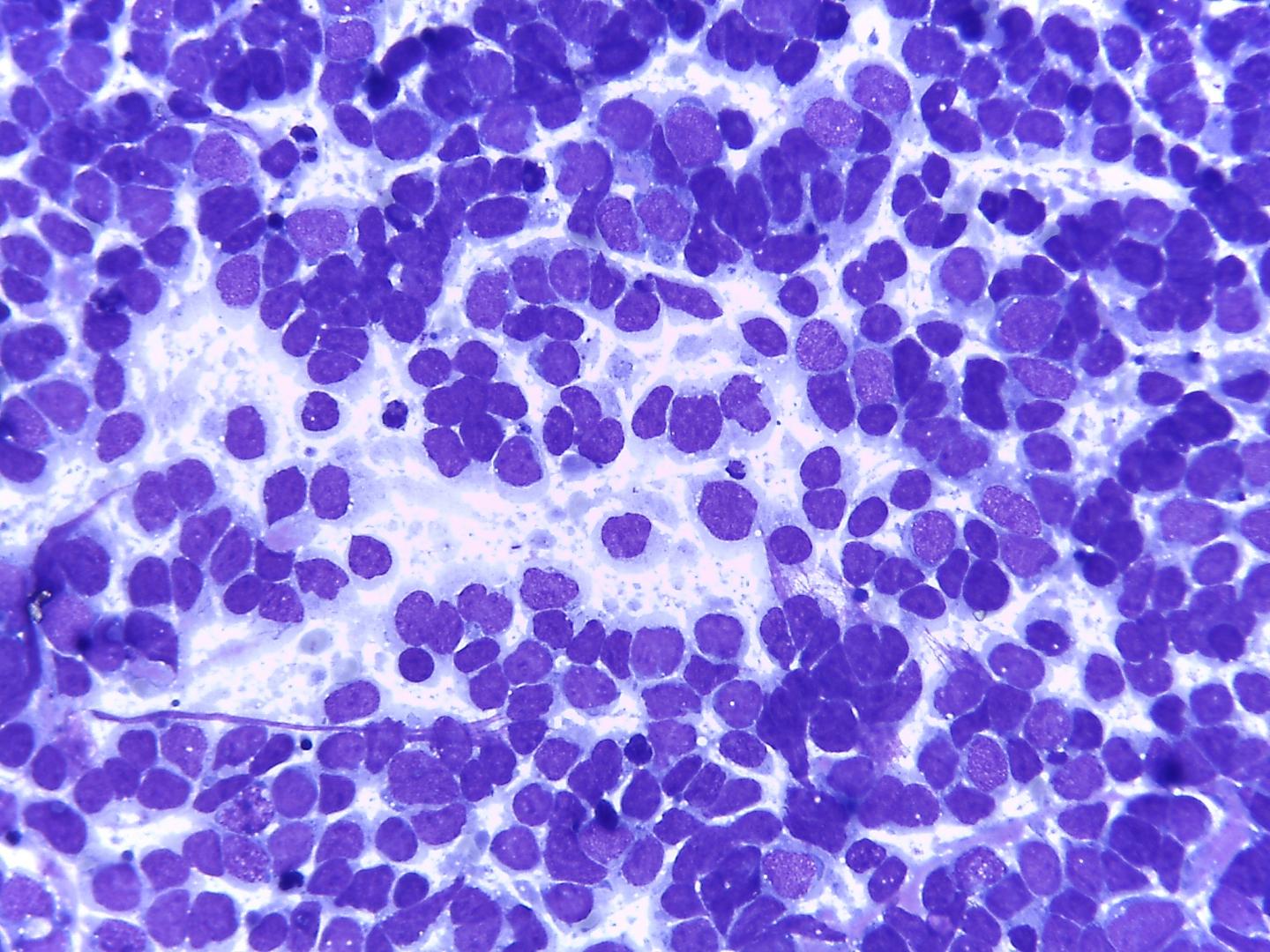
Fig 4a – Wilms ‘tumor-Blastema cells- Loose small round cells with fine chromatin and inconspicuous nucleoli a)-Giemsa- remark nuclei moulding , given the scarcity of cytoplasm
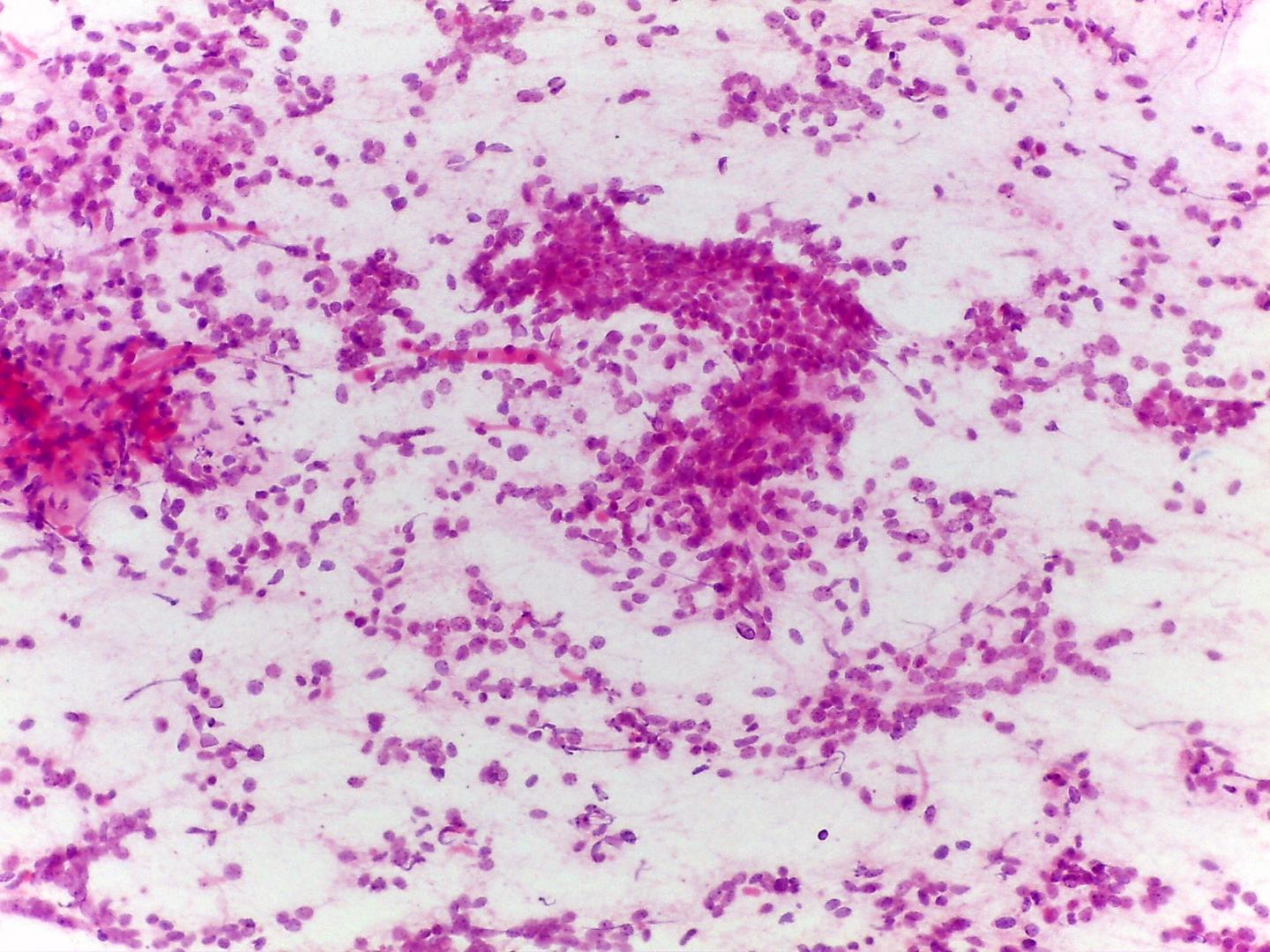
Classically, a triphasic morphologic pattern may be present in differing proportions:
- Blastemal cells (Fig 4a, Fig 4b)
- The most common cells in smears (due to looser cohesiveness they are easy to aspirate).
- Small round blue cells, either singly or in diffuse sheets or rosettes
- Nuclear moulding
- Monotonous pale nuclei with very fine chromatin
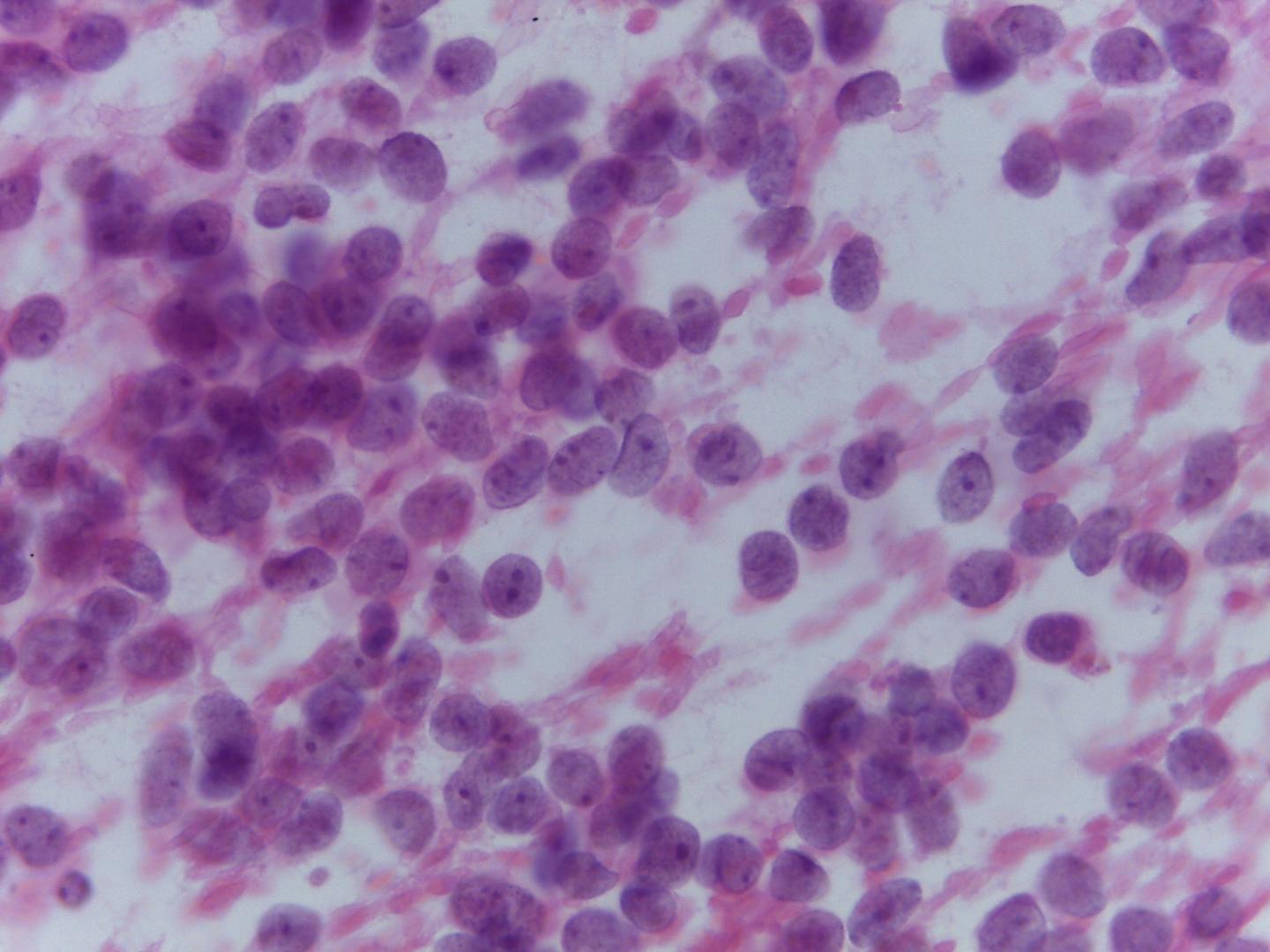
- Stromal component(Fig 5)
- Primitive fibromyxoid stroma
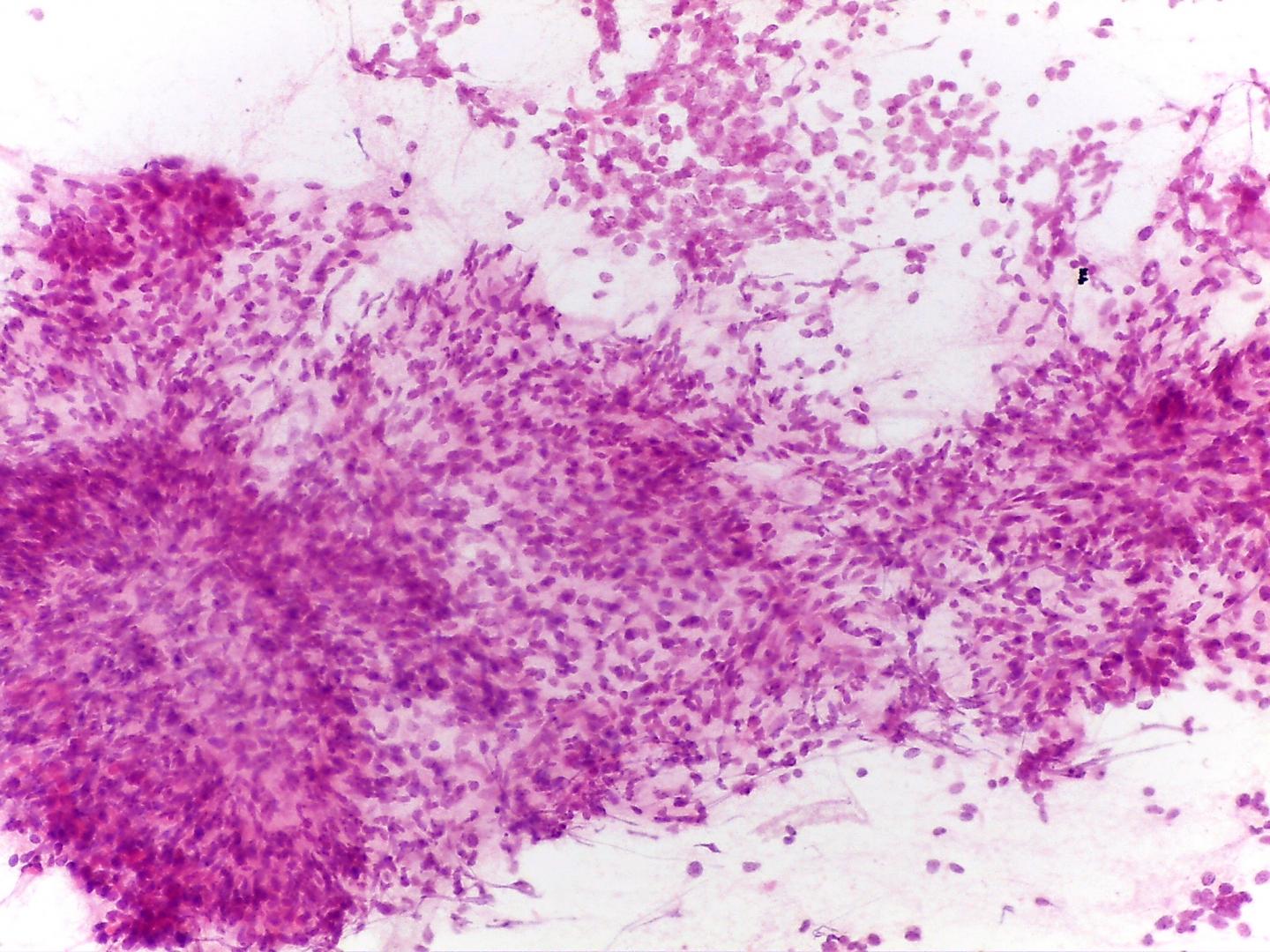
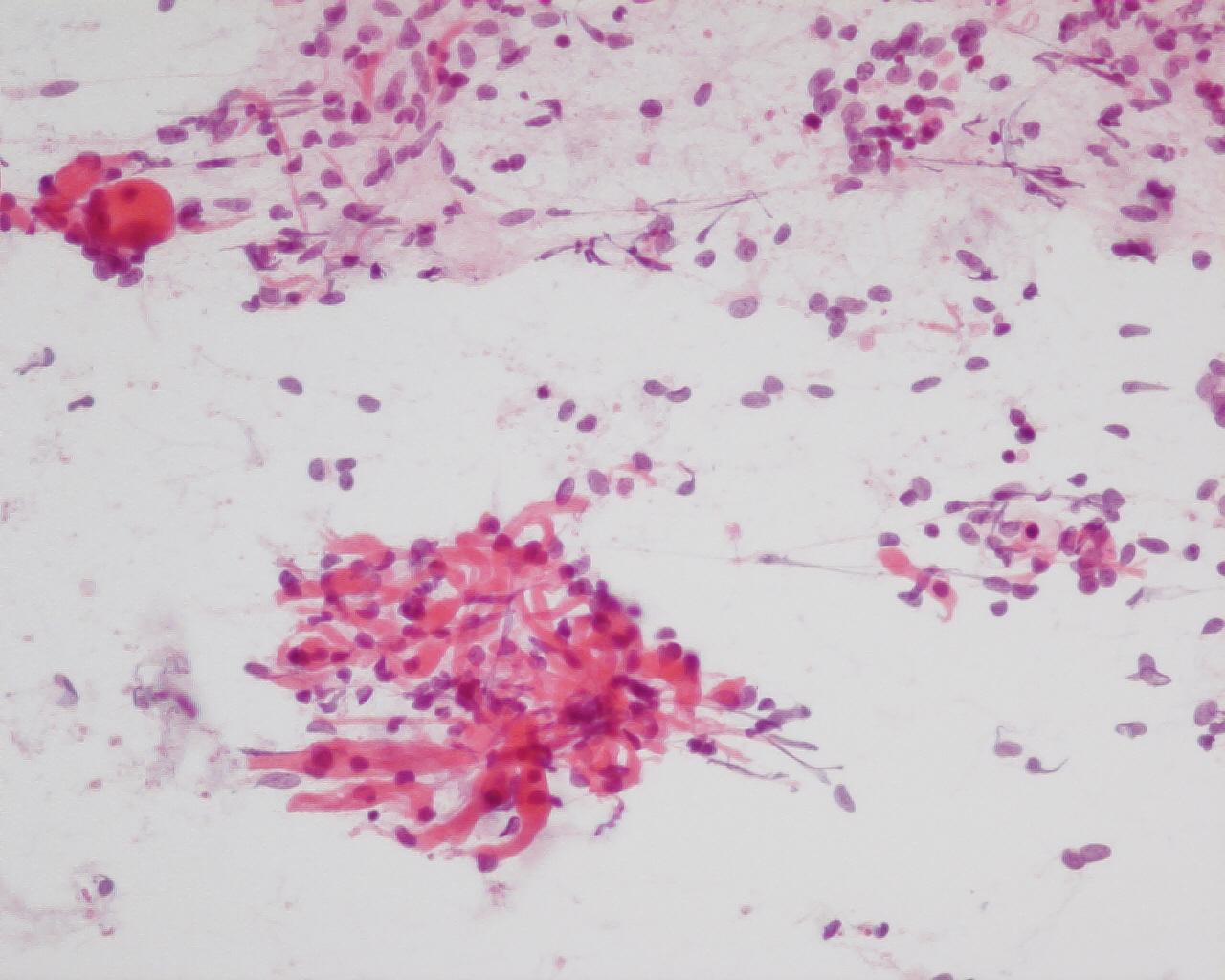
- Rhabdomyoblastic differentiation (Fig 6), chondroid tissue, or any other type of mesenchymal differentiation
- Epithelial cells(.Fig 6)
- Three-dimensional cellular groups, simulating tubules, papillary structures or poorly formed glomeruli
- Cells with high N/C ratio difficult to differentiate from blastemal cells
- Squamous metaplastic cells
- Anaplasia – three criteria must be present:
- Nuclear enlargement (greater than three times the size of other nuclei )
- Hyperchromasia
- Atypical mitotic features
- Present in any of the tumour components
- When present in cytology, it should be considered to be representative of diffuse anaplasia and should be reported
Immunocytochemistry
- Vimentin: positive in blastemal cells
- Wt1: Positive in immature cells (mesenchyma or blastema)
- Bcl2: Positive
- p53: intense and extensive positivity suggests anaplasia
- Cytokeratin: positive in epithelial component
- NSE, synaptophysin, NB84, CD56 N-CAM: in both blastema, mesenchymal and epithelial immature cells)* ,
- Actin and desmin may be positive and are generally of no use in differential diagnosis
Genetic studies:
- 1-2% familial
- 5-10% associated with syndromes and congenital anomalies
- 11p, 1p and 16q deletion/LOH- prognostic relevance
- Two main ways of tumorigenesis have been advanced:
- I- Wnt Pathway
- CTNNB1 and WT1 mutations appear to coexist in some cases of WT (6-20%),
- Association with intralobular nephrogenic rests
- Predominantly stromal type Wilms´ tumours
- Early age onset (1-2 years old)
- Association to WARG or Denys-Drash syndromes
- A novel gene, WTX located at chromosome Xq11.1, is also reported to be mutated in Wilms´ tumours. WTX operates in the AXINAPC complex, down-regulating the activity of beta-catenin
- LOI of IGF2 can be present
- II- IGF2 LOH
- Loss of heterozygoty/imprint of IGF2,
- Associated with perilobular nephrogenic rests
- Association with non-stromal Wilms´ tumours
- Association with Beckwith –Wiedemann syndrome
- Affecting older children (3-4 years old)
- Anaplastic nephroblastoma
- Aneuploidy Mutation of P53
Differential diagnosis
- Neuroblastoma vs blastema
- Monotonous population with small round blue cells
- Neuroblasts in different stages of maturation
- Nuclei with typical salt-and-pepper chromatin
- Fibrillary matrix
- Rosettes with central fibrillary matrix
- Vimentin: usually negative
- Neuroendocrine markers: positive
- Cytokeratin: negative
- Renal PNET vs blastema
- Most common in young adults
- True rosettes
- Nuclei with finely stippled chromatin
- Dark and light nuclei (low power magnification ),(dark nuclei are probably due to apoptosis)
- Nuclei more evenly spaced than blastemal Wilms´ tumour
- Tigroid background (Giemsa stain)
- CD99: positive – (membranous and strong) – (attention must be paid to blastemal cells of Wilms´ tumour that can be positive, even though less strong).
- FLI-1: Positive*- in 90% of the cases
- WT1:Negative ( in most cases)
- *t (11:22) (q24; q12)
- Lymphoma (Burkitt) vs blastema
- Monomorphic population with dispersed cells
- Lipidic cytoplasmic vacuoles
- Typical coarse chromatin
- High number of mitosis and numerous cells in apoptosis
- CD45: positive
- CD10: Positive
- CD38: Positive
- Bcl2: Negative
- Ki-67: 100% positive
- Rhabdomyosarcoma
- Lacks triphasic pattern
- Eccentric cytoplasm
- Differential diagnosis difficult in primitive tumours
- CD56(NCAM): Positive
- Myogenin: positive
- Rhabdoid tumour
- Monomorphous population of large cells with a rhabdoid-like pattern
- Stripped bare nuclei
- Vesicular nuclei with prominent eosinophilic nucleoli
- Eosinophilic Para nuclear cytoplasmic inclusions
- Cytokeratin: positive (dot)
- Vimentin: positive (dot)
- CD10: Positive
- CD56(NCAM): Negative
- INI 1- Loss of expression
- Clear cell sarcoma
- Background with metachromatic mucoid-like material rich in glycoproteins, seen in Giemsa-stained slides
- Stripped cells can simulate blastema
- Neoplastic cells organized in a perivascular pattern (when present, may be helpful)
- Vesicular nuclei with grooving
- WT1: Negative
Main points
- Embryonic neoplasm derived from nephrogenic blastemal cells
- Less than 10% harbour a WT1 mutation
- Constitutional WT1 mutation and Constitutional epigenetic mutation IGF 2 locus are predisponent factors
- It is the only renal tumour of childhood that may be bilateral (5% of the cases) and/or multifocal
- It is the only renal tumour of childhood associated with the presence of nephrogenic residues (30%)
- Diffuse anaplasia
- Present in 5% of the cases
- Not described in children younger than 6 months of age
- More likely in patients older than five years of age
- Represents a more resistant cell line
- Association with extensive and strong immunoreactivity to p53 (p53 mutations)
- Metastasizes to lymph nodes in 15%, or to the lungs, liver or peritoneum
- Rarely metastasizes to the bone (1%)
- Recurrences are more frequent during the first two years after diagnosis